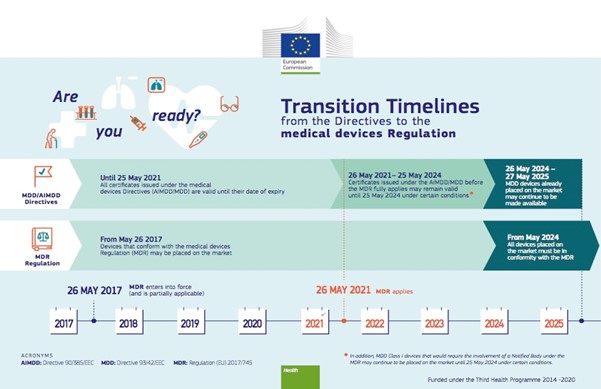
- General medical devices
- Active implantable medical devices
- In-vitro diagnostic medical device
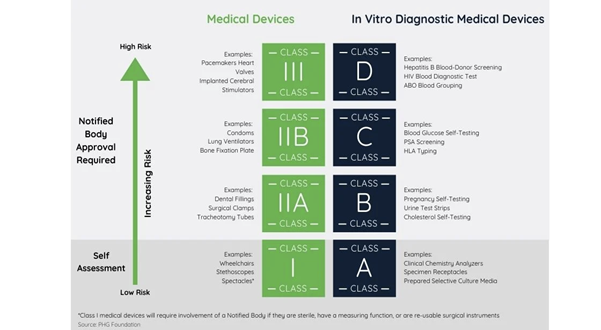
According to MDR Article 51, devices are divided into the following classes I, IIa, IIb and III, considering the intended purpose of the devices and their inherent risks.
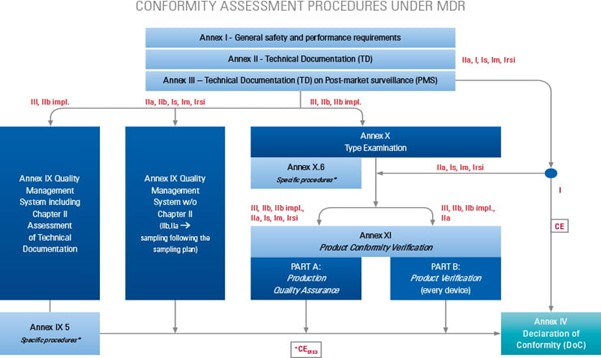
Under MDR a manufacturer must conduct a conformity assessment. This is the process where a manufacturer must demonstrate the requirements of the MDR relating to a device have been fulfilled. Demonstrating conformity is in the first instance the responsibility of the manufacturer.
For the majority of device classifications, the conformity is assessed by a notified body. The higher risk and classification of the device, then the greater the involvement of a notified body in conformity assessment.
A manufacturer must ensure, regardless of the classification of the device, that the general safety and performance requirements are satisfied under MDR Article 5, MDR Annex I.
This includes carrying out the necessary clinical evaluations (MDR Article 5 (3), MDR Article 61, MDR Annex XIV.
For implantable devices and class III devices, a premarket clinical investigation is compulsory, with some exceptions such as modifications of an existing device, demonstrated equivalence to CE-marked device, placed on the market under Directive 90/385/EEC or Directive 93/42/EEC for which sufficient clinical data is already available, and specific exemptions laid down in Article 61(6)(b).